Chronische Lymphatische Leukämie
CLL
Die CLL ist eine leukämisch verlaufende Erkrankung aus der Gruppe der indolenten B-Zell-Lymphome.
Themen im Überblick
Häufigkeit/Inzidenz
Die CLL ist die häufigste leukämische Erkrankung in den westlichen Industrieländern und eine Erkrankung des höheren Alters. Das Durchschnittsalter bei Diagnose liegt bei 70 bis 75 Jahren. Die Inzidenz beträgt bei Männern 7,4 Erkrankungen/100.000 und bei Frauen bei 4,8/100.000 (Wendtner et. al, 2017).
Symptome
Die CLL führt zur lokalisierten oder generalisierten Lymphadenopathie (Lymphknotenschwellung) und Splenomegalie (Milzvergrößerung) mit den entsprechenden Symptomen.
Weiterhin kann es im Verlauf durch die zunehmende Infiltration des Knochenmarkes zu einer hämatopoetischen Insuffizienz mit den daraus resultierenden Symptomen Thrombozytopenie, Anämie und Neutropenie kommen (siehe AML). Diese führen klinisch zu einer variabel ausgeprägten Anämiesymptomatik sowie zu Blutungs- und Infektneigung. Letztere wird häufig durch einen sich entwickelnden Immunglobulinmangel (Antikörpermangel) verstärkt. Weiterhin kommt es bei der CLL häufig zur AIHA (= Autoimmunhämolytische Anämie), AITP (=Autoimmunthrombozytopenie) und anderen Autoimmunphänomenen.
Pathogenese/Entstehungsursachen
Die Ätiologie der CLL ist bisher nicht geklärt. Pathophysiologisch liegt der Erkrankung in der überwiegenden Anzahl der Fälle eine klonale Expansion (krankhafte Vermehrung) CD5+ B-Zellen zugrunde, welche im Verlauf zunächst zu einer Infiltration des Knochenmarkes, später dann auch der Lymphknoten und anderer lymphatischer Gewebe führt (Rytting et al., 2017).
Diagnostik/Klassifikation
Diagnostisch wichtig sind Anamnese, körperliche Untersuchung, Blutbild und Differentialblutbild, Immunphänotypisierung, Abdomensonographie und ggf. auch weitere Parameter (Hämolyseparameter, Immunglobuline) obligat. Eine Knochenmarkpunktion ist zur Diagnosestellung in der Regel nicht erforderlich. Bei Vorliegen einer Therapieindikation sind genetische Untersuchungen (Zytogenetik und Molekulargenetik) indiziert. Die Klassifikation erfolgt nach der Klassifikation von Binet in die 3 Stadien A-C. (Wendtner et. al, 2017).
Chromosomenstatus/Zytogenetik
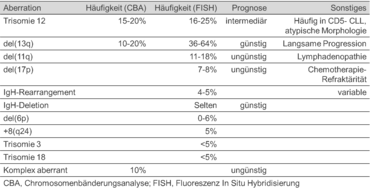
Chromosomenaberrationen lassen sich bei der CLL häufig nachweisen. So finden sich in 50 % aller Patienten, die mittels Bänderungsanalyse untersucht werden und in ca. 80 % aller Patienten, in denen eine FISH-Analyse durchgeführt wird, klonale Anomalien.
Chromosomenaberrationen habe bei der CLL prognostische Relevanz. Die nachfolgende Tabelle zeigt einen Überblick über die häufigsten Anomalien und deren prognostische Bedeutung (Reddy KS., 2005).
Genstatus/Molekulargenetik

Bei ca. 50 % der Patienten mit CLL können somatische Mutationen im Bereich der variablen Region der Immunglobuline nachgewiesen werden. Findet sich hier ein unmutierter Status, ist dies mit einer ungünstigen Prognose assoziiert, die einer speziellen Therapie bedarf. Mit der Einführung des next gerneration sequencing konnten weitere Mutationen bei Patienten mit CLL detektiert und untersucht werden. Die nachfolgende Tabelle gibt hier einen aktuellen Überblick (Ghamlouch et al. 2017).
Die Behandlungsoptionen reichen je nach Stadium, Genetik und Verlauf der Erkrankung von einer „watch and wait“-Strategie bis hin zur allogenen Stammzelltransplantation. Allgemein gilt, dass im Stadium Binet A in der Regel keine Behandlungsbedürftigkeit vorliegt, im Stadium Binet B die Option einer Behandlung geprüft werden sollte und im Stadium Binet C die Therapie indiziert ist.
Aktuelle Leitlinien zur CLL-Therapie finden Sie unter www.onkopedia.com
Literatur
- Wendtner et al., Onkopedia-Leitlinie CLL, Stand Januar 2017
- Rytting et al., MSD Manual, CLL, Ausgabe 2017
- Reddy KS., Chronic lymphocytic leukaemia (CLL) Atlas Genet Cytogenet Oncol Haematol. 2005
- Ghamlouch et al., Chronic lymphocytic leukemia genomics and the precision medicine era. BJH, 2017
Das könnte Sie auch interessieren